ノーザンサイエンスコンサルティング株式会社
ノーザンサイエンスコンサルティング株式会社
ADMET Predictor® は、化合物構造から ADMET 物性を高速・高精度に予測します。第三者による数多くの論文で最も精度が高いとの評価を得ています。また、ユーザーデータに基づく独自の予測モデルを構築するモジュールも準備されており、in silico 初心者からエキスパートまで幅広くご利用いただけます。
2025年6月4日に、新バージョン ADMET Predictor® 13 の日本向けリリースウェビナーが開催されました。ウェビナービデオでご確認ください。
ADMET Predictor® は、化合物の経口吸収に関連する175以上ものADMET物性を分子構造から高精度かつ高速に予測するソフトウェアです。搭載されている物性予測モデル以外に、ユーザーのデータから独自の予測式を構築して利用することも可能です。また、バージョン 8.0 からは、可視化機能やデータマイニング機能が強化され、ケムインフォマティックス領域の利用でも威力を発揮します。化合物の各物性値を比較検討や新規構造の生成、100万を超えるような大規模な化合物ライブラリーの高速スクリーニングと、様々な場面でご利用いただけます。
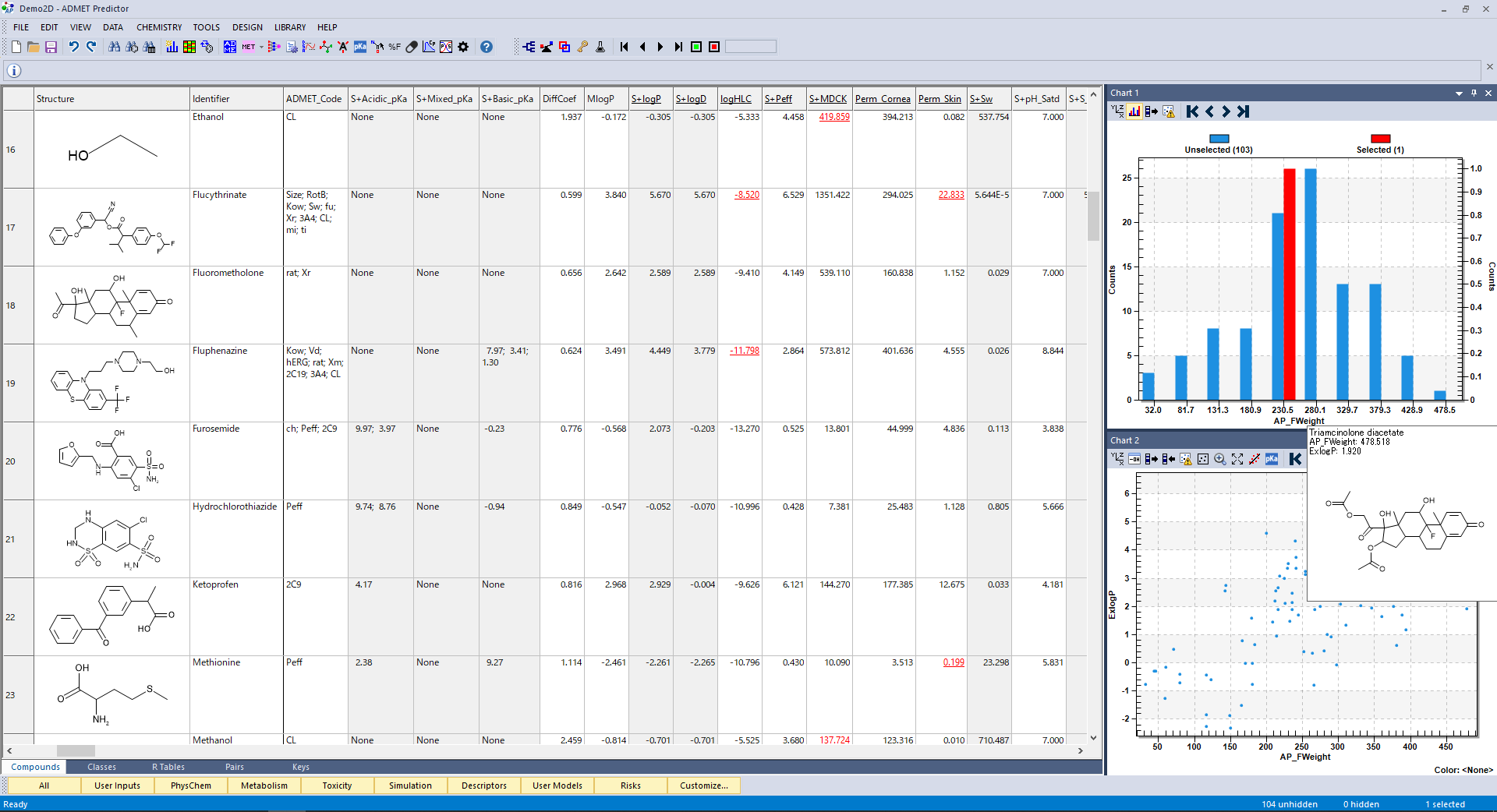
予測モデルは、公的機関や学術文献などから収集した各種物性値の実測値と化学構造を精査し、厳選されたディスクリプターと最適な学習アルゴリズムを用いて構築されています。データの質やディスクリプターの選定、学習アルゴリズムが計算精度と速度に大きく影響するため、長い年月をかけて試行錯誤が繰り返えされ、現在、世界でトップクラスの高精度かつ高速な予測モデルが実現されています。
また、化合物の特性を、独自に吸収、代謝、毒性に関する24の予測プロパティーからスコア化した ADMET Risk でスクリーニングすることができます。さらにヒートマップ表示や最大16種のプロパティーをくさびの長さと色で表現するスタープロットにより、視覚的に化合物の特性を判断することも可能です。
現在、ADMET Predictor は、以下のモジュールで構成されています。
開発元である Simulations Pus 社では、人工ニューラル・ネットワーク(ANN)技術を駆使し、微細イオン化平衡の数量化理論-Microspecies Equilibria を開発しました。この理論と33,000以上の質の高い実測値からの pKa 予測精度は最高レベルとの評価を受けています。
ANN モデル(デフォルト)と Meylan-Howard の方法で予測します。
LennernasとAmidon によるヒトとRatの消化管膜透過性測定値を基に構築したモデルで予測します。
Boehringer や Affymax の公開されているデータを基に構築したモデルで予測します。
High/Low/Undecided で表示と分配係数での表示となります。
370の医薬品のデータから構築したANNモデルで予測します。
331の医薬品のデータから構築したANNモデルで予測します。
GastroPlus® に搭載されているACATモデルをベースに計算します。
EPA の rat estrogen receptor binding データをベースにモデルを構築しました。Tox/Non-Tox/Undecided で表示されます。
化合物構造と毒性と no-effect level(NOEL) の関係を理解するためFDAでは MRTD の Database を構築しています。その Database から作成したモデル式から予測します。経口投与で 3.16mg/kg/day より多いか少ないかの表示となり、少ない場合は副作用の可能性が高いことを表します。
EPA fathead minnow LC50 の Database から構築したモデルで予測します。96時間暴露後の50% 致死率を示す mg/L の数値で表示されます。
Carcinogenic Potency Database (CPDB) のデータを基に発がん性予測モデルを構築しました。CPDB は1200以上の文献及び400以上の米国国立がん研究所の報告書から成る5000を超えるデータを含みます。mg/kg/day の単位で表示されます。
サルモネラ菌10種の菌株での Ames の変異原性試験モデル。Active/Inactive で表示されます。
Tetrahymena Pyriformis(繊毛虫類)急性毒性
文献値をベースにhERG K+チャネルの阻害に係る pIC50 のモデル構築を行いました。医薬品及びドラッグライクな化合物の哺乳類細胞(ヒト胎子腎[HEK]、チャイニーズハムスター卵細胞[CHO]及び心筋細胞)を用いた信頼できる値を採用しています。
ALP、SGOT、SGOP、LDH、GGT の数値上昇。
Simulations Plus社は、Enslein Research Institute と同研究所が保有している Km 値、Vmax 値のデータベースの独占使用契約を締結し、化学構造からヒト CYP 1A2, 2C19, 2C9, 2D6, 3A4 での基質に対する親和性(Km 値)及び最大代謝速度(Vmax)を予測する世界初のモデルを構築しました。Km 値は μM、Vmax 値は nmol/min/nmol P450 及び nmol/min/mg microsomal protein で表示されます。Km/Vamx の予測は、ヒト生理学的 PK/PD モデルでのリスクアセスメントや医薬品開発の意思決定支援にご利用いただけます。
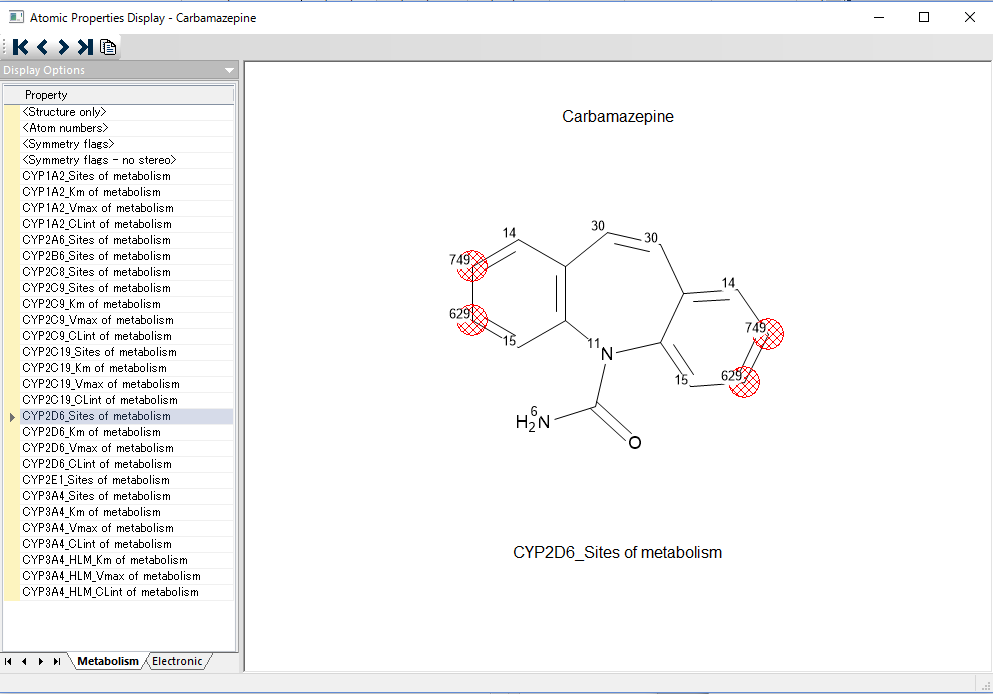
ADMET Predictor X から搭載された Transporters モジュールでは、トランスポーターに特化した一連のモデルが提供されています。
新たに加わった ADMET Predictor Transporters モジュールには、P 糖タンパク質(P-gp)、乳がん耐性タンパク質(BCRP)、有機アニオン輸送ポリペプチド( OATP1B1、OATP1B3)、有機カチオン(OCT1、OCT2)、有機アニオン(OAT1、OAT3)、胆汁酸塩輸出ポンプ(BSEP)の各モデルが含まれています。 ほとんどのトランスポーターについて、基質/非基質および阻害剤/非阻害剤分類モデルと Km 回帰モデルが提供されています。BSEP には、阻害剤/非阻害剤分類モデルと IC50 値を予測する回帰モデルがあります。
このモジュールには、計 24 のモデルが含まれており、そのうち 18 モデルは ADMET Predictor 10.0 (APX) リリースで新たに追加されたものです。これまであったいくつかのモデル (P-gp 基質、P-gp 阻害剤、OATP1B1 阻害剤)についても、更新されたトレーニングセットを使用して再構築されました。
Transporters モジュールでも、マルチコアを利用した計算処理が可能です。すべてのモデルは、特別なライセンスを必要とせずにマルチスレッドモードで動作します。
Simulations Plus 社の別製品 DILIsym(薬物誘発性肝障害(DILI)予測ソフトウェア)内で必要になるインプットパラメータを予測するモジュールです。これらのパラメータは、本来であれば多くの時間と費用をかけ、in vitro 試験から実測値を得る必要がありますが、本モジュールにより容易に in silico 予測値を得ることが可能です。
ミトコンドリア機能不全、活性酸素種(ROS)毒性、MRP3(多剤耐性関連タンパク質3)、BSEP(胆汁酸塩排出ポンプ)、MDR3(多剤耐性タンパク質3)トランスポーターの阻害を予測する 11 のモデルが搭載されており、AP_DILISYMライセンスによってこれらの予測が可能になりますが、2つの BSEP モデルのみは AP_TRANSPORTERS ライセンスがあれば予測可能です。
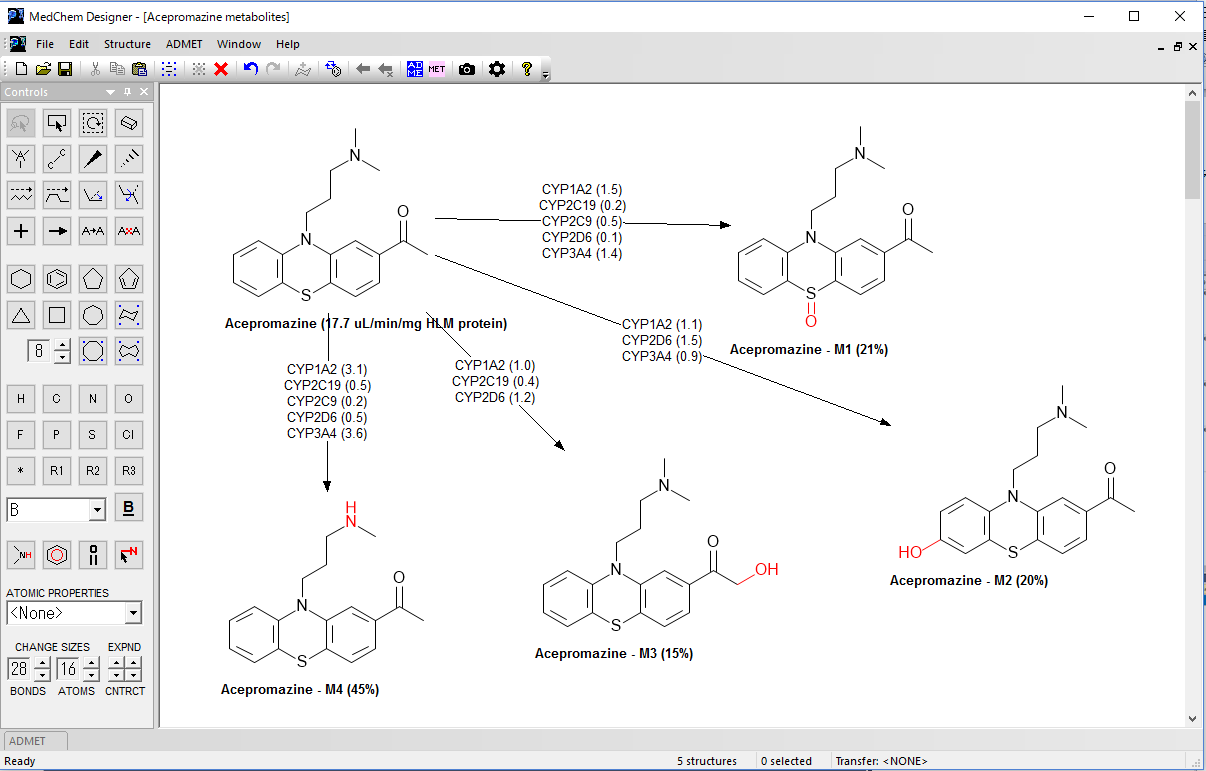
ADMETプロパティ予測値と入力として与えられる投与量を使用し、腸管での吸収率 fraction absorbed(%Fa)、経口バイオアベイラビリティ bioavailability(%Fb)、所定の定常血漿濃度を得るために必要な投与量を推定します。
計算には、GastroPlus® で用いられている Advanced Compartmental Absorption and Transit(ACAT)薬物動態解析システムからの胃および8つの腸コンパートメントが使用されます。それらは、初回通過効果と経口バイオアベイラビリティ(%Fb)の推定のために含まれる肝臓コンパートメントと共に、溶出、透過、輸送、消化管に沿った物理化学的条件(例えば、pH)の変化が考慮されています。全身循環や他の器官は、ひとつの「中心」コンパートメントにまとめられていますが、組織間の組成の変化は、その中心コンパートメントの分布容積(メカニスティックな分布容積、ヒト:S+hVd_PBPK、ラット:S+rVd_PBPK)を計算する際に織り込まれています。
計算結果は、ADMET Predictor のスプレッドシートにカラム追加の形で返されます。
ユーザーインターフェイスは、シミュレーションプロセスを能率的に行うため、できるだけシンプルに設計されています。このため、例えば、メディシナルケミストが腸内吸収やバイオアベイラビリティを素早く評価してみる、といった使い方も容易に実現します。
Simulations Plus 社独自の ACAT モデルを使用して、Rat, Human, Mouse のフィジオロジーについてシミュレーションを実行し、分子構造から予測されたプロパティのみを使用して、化合物の吸収率 (%Fa) およびバイオアベイラビリティ (%Fb) を推定します。
トップのダイアログには現れない LogD や溶解度などの物理化学的特性については、Advaced ボタンからアクセスできる専用ダイアログで設定することが可能です。
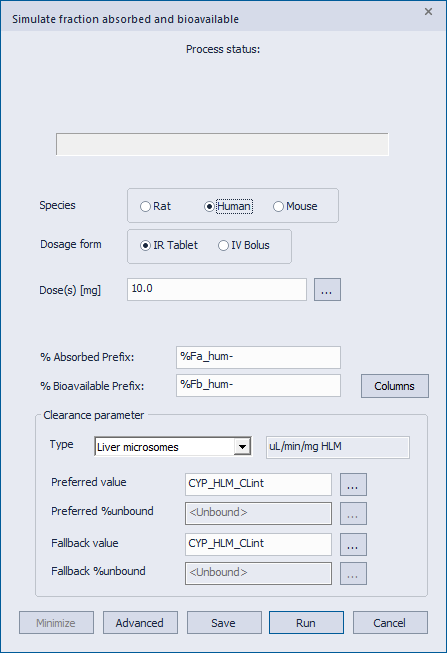
独自の ACAT モデルを使用して HTPK Simulation を実行し、特定の目標血中濃度を達成するために必要な投与量を予測します。上記の %Fa、%Fb のシミュレーションと同様に、3つの Species: Rat, Human, Mouse がサポートされており、一度に1つの種についてのみシミュレーションを行うことができます。
薬物の最小有効投与量に関する情報は、構造活性相関、化合物の優先順位付け、特定の化学物質群の ADMET リスクの同定を推し進める可能性を秘めており、これらの局面における投与量予測に非常に有効であると言えます。
以下の図のように、ダイアログでは、Target Concentration および Initial dosage guess を設定し、Clearance parameters を指定するだけで計算を開始することができます。
構造-物性(QSPR)モデル構築ツールです。化学構造と実測値データセット(物性値や活性値等数値データ)から QSPR 予測モデルの構築が可能です。 モデル構築のプロセスは ADMET Predictor® 8.0 で再設計され、モデル構築パラメータを設定するためのウィザードが準備されています。
2D 構造で 321、3D 構造では 352 ものディスクリプターを発生させます。これらのディスクリプターは、他の QSAR プログラムで利用可能です。ユーザ独自のディスクリプターを用いたり、構造と物性や活性等の数値データからオリジナルのモデルを容易にかつ高速に構築することもできます。
手法:
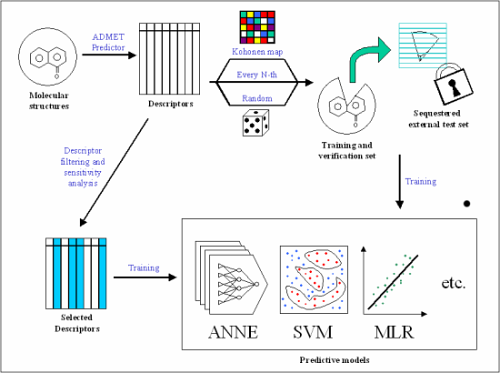
ADMET Predictor には、300を超える原子および分子ディスクリプターが含まれていますが、モデリングアルゴリズムが、低分散および相関するディスクリプターを自動的に削除します。Kohonen マップやK平均法など、いくつかのテストセット選択法が利用できます。テストセットはモデルトレーニングでは使用されないため、外部テストセットを意味しています。最良のモデルを作成するのに必要なディスクリプターおよびニューロンの数は、先験的には分かっていないため、異なるアーキテクチャのグリッド、すなわち異なる数のニューロンおよびディスクリプターを作成することになります。最適なモデルを選択するアルゴリズムもいくつか備わっています。
AIDD モジュールは、化合物の多目的最適化を可能にする AI Drug Design 機能と、様々な手法による新規構造デザインを可能にする Compound Design 機能があります。
ADMET Predictor の高精度な ADMET 物性予測モデルに、多目的な化合物最適化の機能が統合されたものです。
1 つ以上の開始構造を取り、一連のターゲットプロパティに対してそれらを最適化します。すべての数値 ADMET 物性予測モデルおよびカスタムモデルは、ターゲットプロファイルの一部として用いることができます。これには、HTPK シミュレーションモジュールのメカニズムモデル(%Fa および %Fb)が含まれます。
また、バージョン 10.3 からは、ドッキングプログラム、Python または R スクリプトなどの外部アプリケーションで計算された予測値も指定できるようになりました。各最適化サイクル中に、外部アプリケーションが自動的に呼び出されて、生成された新しい化合物の計算を行います。
以下のワークフローにより、化合物の最適化を行います。
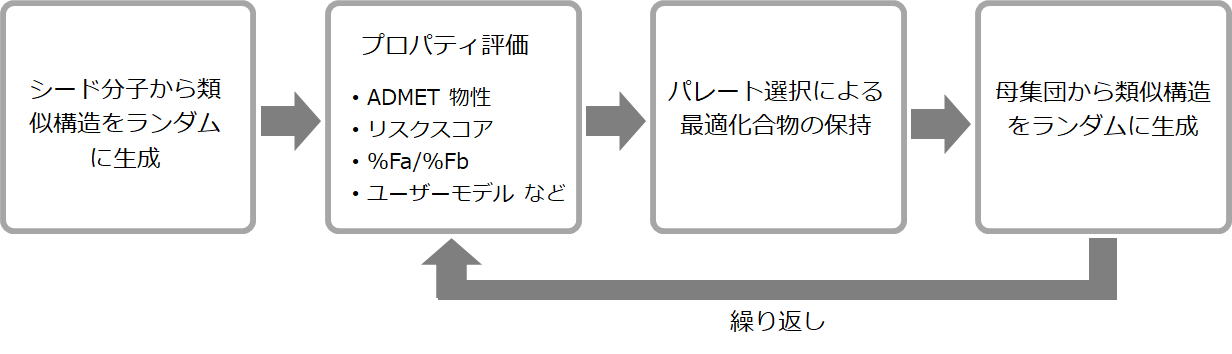
AI Drug Design では、現在利用可能なすべての ADMET 物性予測モデル、他のカスタムグローバルモデル、ローカルモデル、またはデルタモデルにおいて、マルチスレッドモードが利用可能です。これらの処理機能を活用し、目的の化合物または骨格構造周りのケミカルスペースの大部分を探索するような計算処理で、一晩で、最大 500 万個の分子の生成と評価を実現します (8 コアラップトップコンピューター)。
最適化の一部として、分子のどの部分が変更され、どの部分が保持されるべきかをケミストがコントロールすることが可能です。また、置換基を適用できる位置を指定し、合成の実現可能性制約またはターゲットに関する事前の知識に基づいて生成される化合物を制御することもできます。
ADMET Predictor の分析ツールでは、計算結果をフィルタリングし、分析する機能が提供されています。最終結果を調べて、どの化合物が物性の最適な組み合わせを持っているかを知ることができます。また、計算の進行中であっても、各生成サイクルの中間結果を分析することも可能です。
以下の手法により新しい化合物を生成することができます。
反応スキームと反応物リストを使用
ひとつ以上の構造から開始して複数の構造変換ルールを使用
R アタッチメントを持つスキャフォールドと置換基リストを使用
R テーブルを使用し、既存の置換基を組み合わせた化合物を生成
SMIRKS(SMK)、MDL 反応(RXN)、CRF で定義された反応を化合物に適用し、スキャフォールド、R グループの置換操作で使用するフラグメントファイルを生成
置換基をフラグメントファイルから抽出した代替物に置換
スキャフォールドをフラグメントファイルから抽出した代替物に置換
バージョン 10.3 から、MedChem Studio モジュールの以下のケムインフォマティクス機能が ADMET Predictor の基本機能として利用できるようになりました。データマイニングや化合物データベース作成を行うための機能が搭載されており、創薬初期フェーズでのリードの同定などに役立ちます。
クラス生成技術は、共通構造をベースにケミストが考えるように分類します。
R Tables タブをクリックするだけで自動的にスキャホールドとテーブルを生成します。各R基のカラムが生成されます。
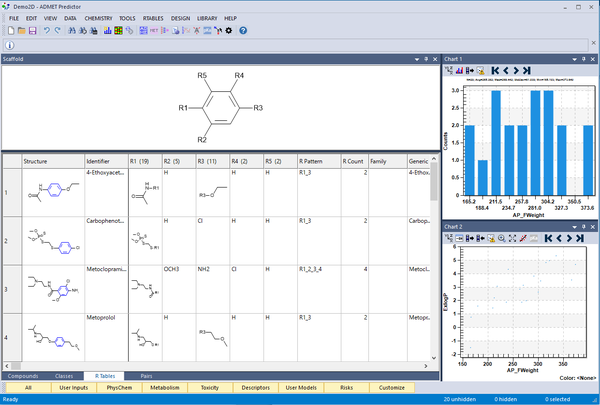
一つを除いて同じ置換基のファミリーカラムを作ることができます。これにより、置換基の変化による活性の変化を解析することができます。
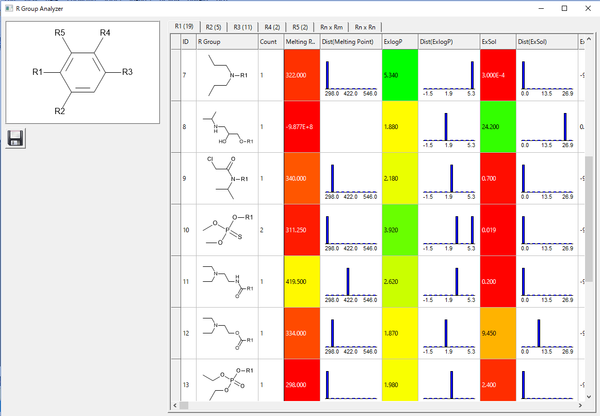
各R基の位置にあるユニークな置換基を表示させ、分子プロパティーを解析できます。 分子プロパティーの平均値や分布を表示させることで、どの置換基が有効なのかを判別するのに有用です。
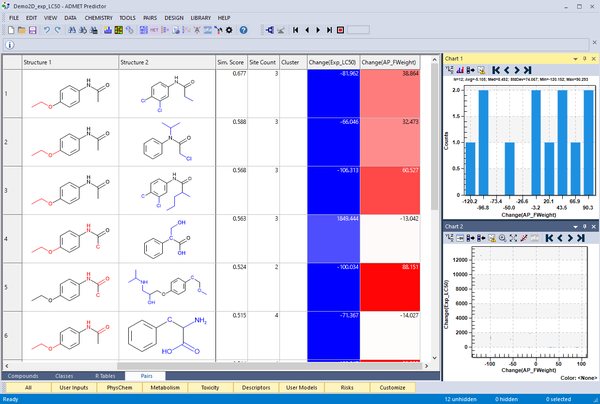
類似性検索を行い、Pairタブでヒットを分析することができます。クエリーとヒット分子は、共通部分構造と異なる箇所が赤と青に表示された分子によって、整列表示されます。
検索をスピーディーにできるように独自の化合物(ライブラリー)のデータベースを作ることができます。 ライブラリーは、構造、テキスト、または類似性で選別できます。 類似性検索は、単一分子や構造ファイルで行うことができます。
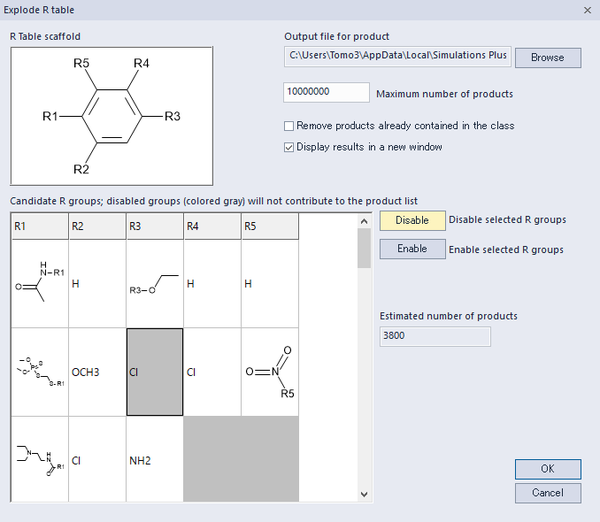
R Group分析で空のカラムは、ケミカルスペースがわからないことを示しています。R Table explosionは、各位置の各置換基と他すべての位置の置換基の組み合わせを行い、新たな分子を発生させることができます。また、特定の置換基を省いて、分子を生成させることもできます。
Matched molecular pair analysis は、データセット中の activity cliff の同定や構造とプロパティーの傾向を見ることができます。
 |
GastroPlus® 薬物動態解析・製剤設計支援ソフトウエア |
 |
ADMET Predictor® ADMET 物性予測ソフトウエア |
 |
DDDPlus™ in vitro 溶出試験シミュレーションソフトウエア |
 |
MedChem Designer™ 分子構造作成と ADMET 物性予測機能を備えたフリーソフト |
 |
MembranePlus™ 膜透過性試験データをベースにしたシミュレーションツール |
©2004-2026 Northern Science Consulting Inc. All Rights Reserved.

 ADMET Predictor 製品資料
ADMET Predictor 製品資料